You will get advanced protein-ligand docking simulations to drug discovery
Top Rated
Top Rated
Project details
The services include:
STARTER "Essential Docking Analysis":
Basic semi-flexible protein-ligand (native + new) docking at the active site.
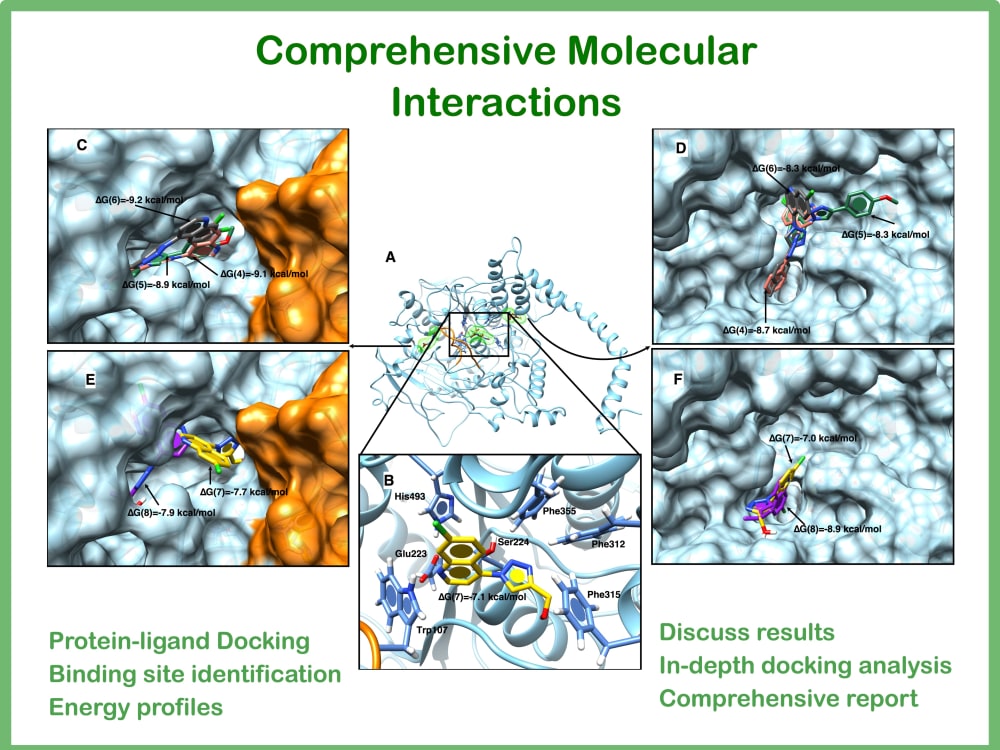
Energy profiles and general intermolecular interaction analysis.
One amazing and high-quality 3D (publication style).
STANDARD "Advanced Docking Dynamics":
AI-based flexible protein-ligand (native + new1) docking at the active site
Energy profiles and intermolecular interaction analysis.
Two amazing and high-quality 3D (publication style).
ADVANCE "Comprehensive Molecular Interaction":
AI-based flexible protein-ligand (native + new1 + new2) docking at the active site
In-depth docking analysis (Energy and intermolecular interactions) + 30 minutes to explain results.
Three amazing and high-quality 3D - 2D interaction analysis images (publication style).
Comprehensive interaction report.
Software: AutoDock Vina, SwissDock, Blotz, RosettaDock, PyMOL, VMD, UCSF Chimera.
This project will aid in understanding molecular interactions and guide drug design with high precision. Ideal for researchers and professionals in the first step of drug design who require precise modeling of protein-ligand interactions.
STARTER "Essential Docking Analysis":
Basic semi-flexible protein-ligand (native + new) docking at the active site.
Energy profiles and general intermolecular interaction analysis.
One amazing and high-quality 3D (publication style).
STANDARD "Advanced Docking Dynamics":
AI-based flexible protein-ligand (native + new1) docking at the active site
Energy profiles and intermolecular interaction analysis.
Two amazing and high-quality 3D (publication style).
ADVANCE "Comprehensive Molecular Interaction":
AI-based flexible protein-ligand (native + new1 + new2) docking at the active site
In-depth docking analysis (Energy and intermolecular interactions) + 30 minutes to explain results.
Three amazing and high-quality 3D - 2D interaction analysis images (publication style).
Comprehensive interaction report.
Software: AutoDock Vina, SwissDock, Blotz, RosettaDock, PyMOL, VMD, UCSF Chimera.
This project will aid in understanding molecular interactions and guide drug design with high precision. Ideal for researchers and professionals in the first step of drug design who require precise modeling of protein-ligand interactions.
Data Tool
PythonWhat's included
| Service Tiers |
Starter
$50
|
Standard
$100
|
Advanced
$150
|
|---|---|---|---|
| Delivery Time | 1 day | 2 days | 4 days |
Number of Revisions | 1 | 1 | 2 |
Number of Graphs/Charts | 2 | 2 | 3 |
Number of Variations | 1 | 1 | 1 |
Data Source Connectivity | - | - | - |
Web Embedding | - | - | - |
Interactive/Animated Visuals | - | - | - |
7 reviews
(6)
(1)
(0)
(0)
(0)
This project doesn't have any reviews.
EE
Eleanor E.
Jun 17, 2026
Biophysical chemistry tutor needed (College/university student)
Very knowledgeable and helpful
RD
Ryan D.
Dec 26, 2025
Professional computation chemist to evaluate two chemical structures.
I had an overall good experience working with Mario on this hectic project. He was very knowledgeable of the areas of this project, very professional and tackled challenges that arose effectively and efficiently. Communications were good as was maintenance of schedule. I will look him up if I have similar work to be done in future.
EE
Eleanor E.
Nov 17, 2025
Biophysical chemistry tutor needed (College/university student)
Very patient, very thorough, excellent teacher! Could have not gotten through physical chemistry without him!
WR
Will R.
Aug 20, 2025
Molecule Design Specialist Needed
very nice work designing in silico and in vitro experiments and molecule designs
MF
Megan F.
Apr 11, 2025
Need images/figures of 2D/3D micellular molecule complex
Mario is very knowledgeable and collaborative. He really understood the science of my project and I learned a lot from our conversations.
About Mario A
Ph.D. in Chemistry - Advanced Computational Biophysics & Drug Design
100%
Job Success
Bucaramanga, Colombia - 11:58 am local time
More than 20 years as a professor and researcher.
Teaching various university-level courses such as Physical Chemistry, Biochemistry, Molecular Biophysics, and Quantum Chemistry (undergraduate and postgraduate levels).
My research, often at the forefront of innovation, has been instrumental in the field, focusing on drug design; insecticide development, larvicidal activity studies, and more, with several notable publications to my credit.
20 years as a consultant researcher. Understanding the industrial process at a molecular level and designing materials.
I have a profound command of molecular modeling software like GAUSSIAN, GAMESS, MOPAC, PySCF, GROMACS, NAMD, LAMMPS, AMBER, AutoDock, Vina, ZDock, HADDOCK; visualization software like VMD, Chimera, PyMOL, Ovito, MarvinSketch, and a suite of bioinformatics tools (ADMEtox, molmin), which have been central to my research endeavors. These include evaluating, predicting, and understanding (Analysis):
- biomolecular & bimaterial structures
- protein-ligand docking & interactions
- protein-protein docking & interactions
- antigen-antibody docking & interactions
- protein-nucleic acid docking & interactions
- Spontaneity of molecular processes
- Molecular Dynamics simulations
...contributing significantly to both academic and industry applications.
Python coding expertise for the extraction of properties of compounds, as well as the training and utilization of AI (artificial intelligence) models for the prediction of compounds in the context of drug design (Computer-Aided Drug Design - CADD). Also used deep generative models (machine learning) for the discovery of new molecules (drug discovery) and materials.
My skill set extends beyond academic research. I am proficient in various operating systems (Linux, macOS, Windows) and adept at scripting for seamless simulation program operations. Even during challenging times, such as the COVID-19 pandemic, my expertise in remote molecular simulation research ensured uninterrupted, high-quality results.
I offer my deep-rooted knowledge and innovative approach to solving complex molecular and material design challenges, bridging the gap between microscopic processes and macroscopic applications in academic and industrial settings.
Let's collaborate to advance your project with cutting-edge scientific insights and solutions!!
Steps for completing your project
After purchasing the project, send requirements so Mario A can start the project.
Delivery time starts when Mario A receives requirements from you.
Mario A works on your project following the steps below.
Revisions may occur after the delivery date.
Initial data
Client shares the protein structure (PDB, CIF or FASTA) and compound structures (SMILES/PDB/MOL/SDF/etc).
Molecular docking
I will run molecular docking simulations and interaction analysis.